利用排序后的bam文件计算基因表达量counts
1、安装subread,其中包括 featureCounts 工具。
11conda install -c bioconda subread2、利用featureCounts计算外显子exon表达量。
311# 定义要循环的路径列表3paths=(4 "/home/hztext/dai/Ana_trans/bam_file/sorted/Tc_Female"5 "/home/hztext/dai/Ana_trans/bam_file/sorted/Tc_Fruit"6 "/home/hztext/dai/Ana_trans/bam_file/sorted/Tc_Male"7 "/home/hztext/dai/Ana_trans/bam_file/sorted/Tc_Petals"8 "/home/hztext/dai/Ana_trans/bam_file/sorted/Tc_VegOr"9)10#定义输出路径11output_path="/home/hztext/dai/copy_Ana_trans/counts_file"12mkdir -p "$output_path"13
14
15# 循环遍历路径列表16for path in "${paths[@]}"; do17 # 在每个路径下执行您的命令18 folder_name=$(basename "$path")19 cd "$path" # 切换到当前路径20#指定基因组注释文件路径和文件名21annotation_file="/home/hztext/dai/Ana_trans/Toona_cilita_var_Genome/Toona_ciliata_LG_Genome.gff"22
23# 循环遍历 BAM 文件24for bam_file in "$path"/*.bam; do25 # 提取文件名作为样本名26 sample_name=$(basename "$bam_file" .bam)27
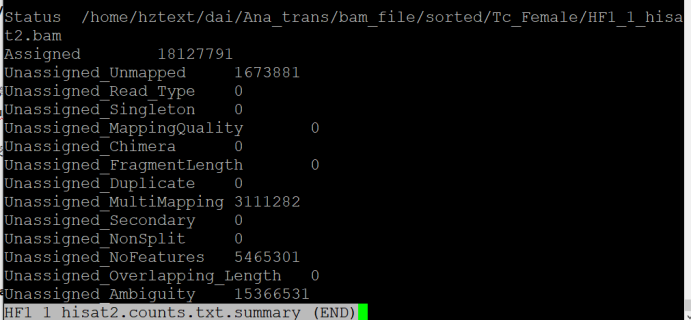
28 # 使用 featureCounts 计算表达量。-T:指定线程数;-a:指定注释文件;-o指定输出路径(不填则为工作路径)/文件名;-p:指定双端测序;-t:指定输出的表达量是mRNA,gene或者exon等;-g:基因标识符属性(如'ID=Ts_Contig1_G00001.t1.exon1;Parent=Ts_Contig1_G00001.t1'则为ID)29 featureCounts -T 50 -a "$annotation_file" -o "$output_path"/"${sample_name}.counts.txt" -p -t mRNA -g ID "$bam_file"30done31done
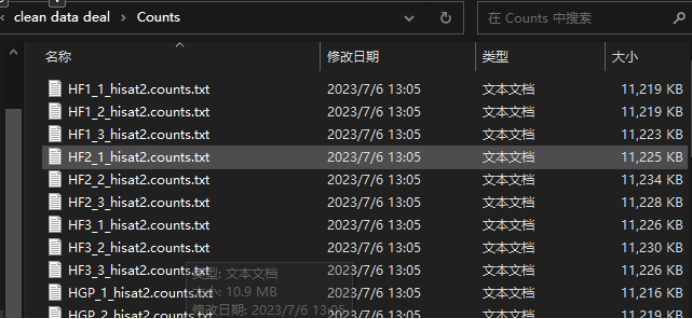
3、表达量counts文件整理
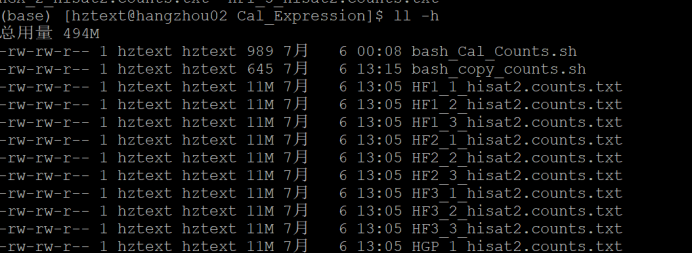
将各文件夹的count.txt文件统一复制到Cal_Expression文件夹内,方便后续下载到电脑。
将以下代码复制并新建bash_copy_counts.sh文件,用bash bash_copy_counts.sh命令运行,结果如下:
211# 定义要循环的count.txt文件现存的路径列表3paths=(4 "/home/hztext/dai/Ana_trans/bam_file/sorted/Tc_Female"5 "/home/hztext/dai/Ana_trans/bam_file/sorted/Tc_Fruit"6 "/home/hztext/dai/Ana_trans/bam_file/sorted/Tc_Male"7 "/home/hztext/dai/Ana_trans/bam_file/sorted/Tc_Petals"8 "/home/hztext/dai/Ana_trans/bam_file/sorted/Tc_VegOr"9)10
11# 循环遍历路径列表12for path in "${paths[@]}"; do13 # 在每个路径下执行您的命令14 cd "$path" # 切换到当前路径15
16# 遍历每个 Counts.txt 文件17for counts_file in "$path"/*.txt; do18#将$counts_file文件复制到/home/hztext/dai/Ana_trans/Cal_Expression路径中。19 cp $counts_file /home/hztext/dai/Ana_trans/Cal_Expression20done21done4、利用WinSCP软件将所有count.txt文件下载到个人电脑。