不同样本基因差异表达分析
利用Rstudio新建一个名为Differential_Expression_analysis的project,利用以下代码进行分析。仅需修改tpm_input的路径、样本名conditions0 。
xxxxxxxxxx1#清空当前环境中的所有变量2rm(list = ls())3{4
5 #如果没有安装"BiocManager",则进行安装,否则跳过6{if (!requireNamespace("BiocManager", quietly = TRUE))7 install.packages("BiocManager")8}9#安装并加载R包"DESeq2"10{if (!requireNamespace("DESeq2", quietly = TRUE))11 BiocManager::install("DESeq2")12}13
14library(DESeq2)15#输入TPM数据16tpm_input <- read.table(file = "G:/R-Demo/counts_to_TPM/all_TPM.txt",17 header = T,row.names = 1,sep = "\t")18#去除各转录组中tpm值均为0的基因19tpm <- tpm_input[rowSums(tpm_input) != 0,]20# 将TPM数据乘以1000并四舍五入,不保留小数21tpm <- round(tpm)22head(tpm)23
24# 创建条件信息:需要因子factor格式25#主要是转录组中包含重复,需要指定哪些重复是对应一个样本的,后续需要对不同样本进行比较26conditions0 <- c("HF1", "HF2", "HF3", "HGP", "HGX", "HL", "HM1", "HM2",27 "HM3", "HP1", "HP2", "HP3", "HR", "HS", "HZZ")28
29# 嵌套循环比较两两条件30for (i in 1:(length(conditions0)-1)) {31 for (j in (i+1):length(conditions0)) {32 condition1 <- conditions0[i]33 condition2 <- conditions0[j]34
35 # 打印两个条件的比较结果36 print(paste("Comparing", condition1, "and", condition2))37 38 # 在这里执行比较的操作,根据具体需求进行相应的处理39#将需要比较的对象与数据中的各列一一对应40conditions <- as.factor(rep(c(condition1, condition2),each = 3))41#整理countData:将两者数据分别进行查找录入42{43 tpm_DES1 <- tpm[,grep(condition1, colnames(tpm_input))]44 #新增一列Geneid进行合并45 tpm_DES1$Geneid <- rownames(tpm_DES1)46 tpm_DES2 <- tpm[,grep(condition2, colnames(tpm_input))]47 tpm_DES2$Geneid <- rownames(tpm_DES2)48 #将两个数据框进行合并,以Geneid为参考49 tpm_DES <- merge(tpm_DES1,tpm_DES2, by = "Geneid")50 rownames(tpm_DES) <- tpm_DES[,1]51 #去除Geneid列52 tpm_DES <- tpm_DES[,-1]53 #将在各转录组中均为0的基因去掉54 tpm_DES <- tpm_DES[rowSums(tpm_DES) != 0,]55}56# 创建 DESeq2 的数据对象57#countData是表达量矩阵或者数据框58#colData是使用condition作为差异表达分析的条件变量。59#在这种设计下,DESeq2将计算每个基因在不同条件下的差异表达。60dds <- DESeqDataSetFromMatrix(countData = tpm_DES,61 colData = data.frame(condition = conditions),62 design = ~ condition)63
64#进行差异分析:这一步耗时较长,45个转录组的count文件花费大约2小时65dds <- DESeq(dds)66#获取差异表达基因的统计结果。67res <- results(dds, cooksCutoff = F, independentFiltering = F)68
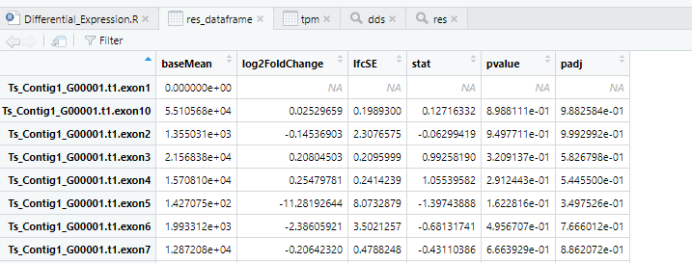
69#查看前几行70head(res)71#将res转成dataframe格式72res_dataframe <- data.frame(res)73head(res_dataframe)74#如果没有安装dyplr,则安装dyplr,否则会直接跳过75{if (!requireNamespace("dplyr", quietly = TRUE))76 install.packages("dplyr")77}78#加载dplyr包79library(dplyr)80#在res_dataframe中新建group列81#根据log2FoldChange和padj条件进行写入“up”或者“down”,将结果赋予res_dataframe282res_dataframe %>%83 mutate(group =case_when(84 log2FoldChange >= 2 & padj <= 0.05 ~ "UP",85 log2FoldChange <= -2 & padj <= 0.05 ~ "DOWN",86 TRUE ~ "NOT_Change"87)) -> res_dataframe288#统计group列中不同数值的情况89table(res_dataframe2$group)90#输出路径定义:91File_output_path <- "./Differential_Expression_results/"92#输出文件名定义:93File_name <- paste0(condition1, "_vs_", condition2,".csv")94
95output_file <- paste0(File_output_path, File_name)96#导出.csv文件,quote=F:禁止在文本中添加“”97write.csv(res_dataframe2,file = output_file, quote = F)98
99#绘制火山图100#加载R包ggplot2101library(ggplot2)102#可选:筛选-log10(padj)>200 且<300的基因名103{104res_dataframe2$padj200 <- (-log10(res_dataframe2$padj))105#筛选上调UP基因106padj200_UP <- res_dataframe2[res_dataframe2$padj200 > 100107 & res_dataframe2$group == "UP",]108#筛选下调UP基因109padj200_DOWN <- res_dataframe2[res_dataframe2$padj200 > 100 110 & res_dataframe2$group == "DOWN",]111#要实现标签尽可能不重叠,可以利用ggrepel包进行修改112{if (!requireNamespace("ggrepel", quietly = TRUE))113 install.packages("ggrepel")114 library("ggrepel")115}116
117}118
119#设置数据来源,一般要求是dataframe格式,x轴是log2FoldChange,y轴是经过-log10()转化的padj120plot <- ggplot(res_dataframe2,aes(x=log2FoldChange,y=-log10(padj)))+121 #设置绘制类型为“散点”图,标签为group列的三个类别122 geom_point(aes(color = group))+123 #手动设置类别的填充颜色124 scale_color_manual(values = c("dodgerblue","gray","firebrick"))+125 #若要显示-log10(padj)>200 且 <300的基因名,则运行这一段代码。126 #max.overlaps:避免标签重叠;size:设置标签大小127 #上调基因标签128 geom_label_repel(data = padj200_UP, aes(x=log2FoldChange,y=-log10(padj)),129 label= rownames(padj200_UP), color= "lightpink",130 max.overlaps = 23, size =3)+131 #下调基因标签132 geom_label_repel(data = padj200_DOWN, aes(x=log2FoldChange,y=-log10(padj)),133 label= rownames(padj200_DOWN), color= "skyblue",134 max.overlaps = 23, size =3)+135 136 theme_bw()137plot_output_path <- "./Plots_output/"138plot_name <- paste0(condition1, "_vs_", condition2,".pdf")139
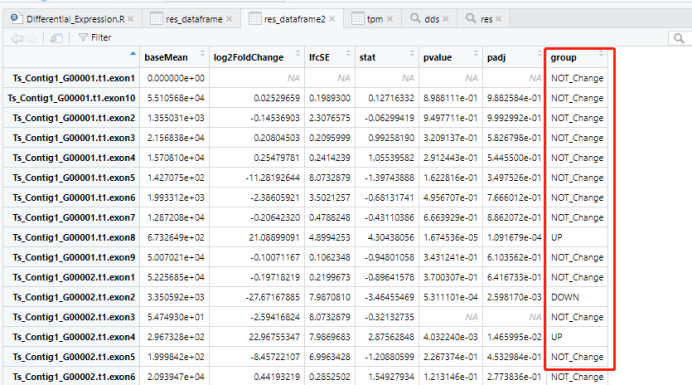
140output_plot <- paste0(plot_output_path,plot_name)141ggsave(filename = output_plot, plot = plot,width = 14, height = 14, units = "in", dpi = 600)142{143Finished_file <- length(list.files(File_output_path, pattern = "\\.csv$", full.names = TRUE))144Finished_plot <- length(list.files(plot_output_path, pattern = "\\.pdf$", full.names = TRUE))145# 比较完成后进行下一对条件的比较146print(paste0(condition1," vs ",condition2," Comparison complete"))147print(paste0(Finished_file," files had completed"))148print(paste0(Finished_plot," plots had completed"))149#打印已完成的比较组合150print(sub("\\.csv$","",list.files(File_output_path, pattern = "\\.csv$", full.names = F)))151} 152}153}154}附表1:差异基因统计结果
附表2:统计group列中不同数值的情况,即上调、下调或没有差异表达